2025-08-01
Precision medicine, pharmaceutical development, biotechnology, and genomic research have all advanced under a framework that classifies variants into fixed categories such as pathogenic or benign. This was a necessary compromise, adopted when reference data were limited and statistical infrastructure immature. But categorical labels collapse uncertainty and obscure quantitative reasoning.
With a massive scale of genomics and refernce data now available, we can return to the foundational tools of probability theory and statistical inference, methods originally developed to address uncertainty in biological. These same principles underpin modern AI, bioinformatics, and statistical genetics. The correct frame of reference provides a coherent framework for replacing categorical classification with rigorous, probabilistic interpretation of variant effect.
We demonstrate this approach with a simplified example. Imaging a human who is suffering from a rare genetic disease. An autosomal dominat autoinflammatory condition, potentially caused by a gain-of-function variant in a gene that is known to cause this condition.
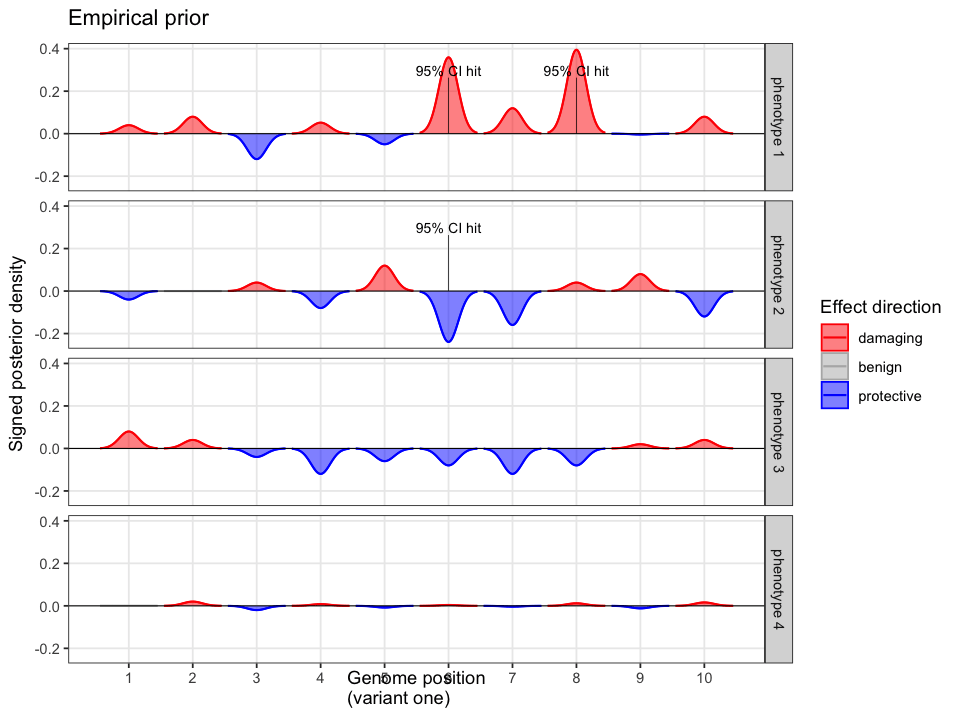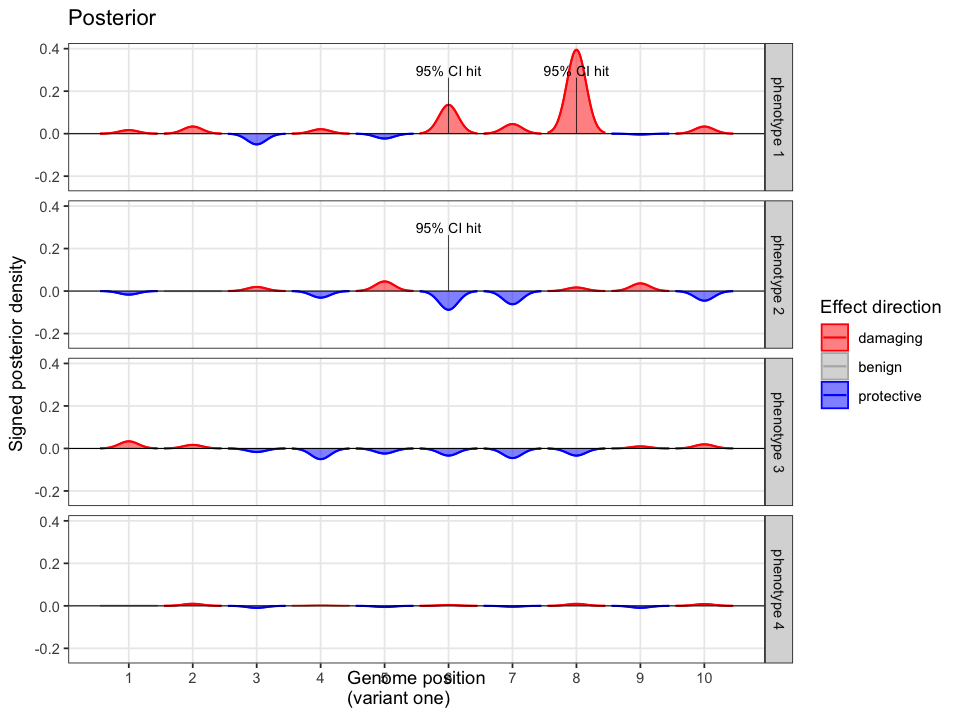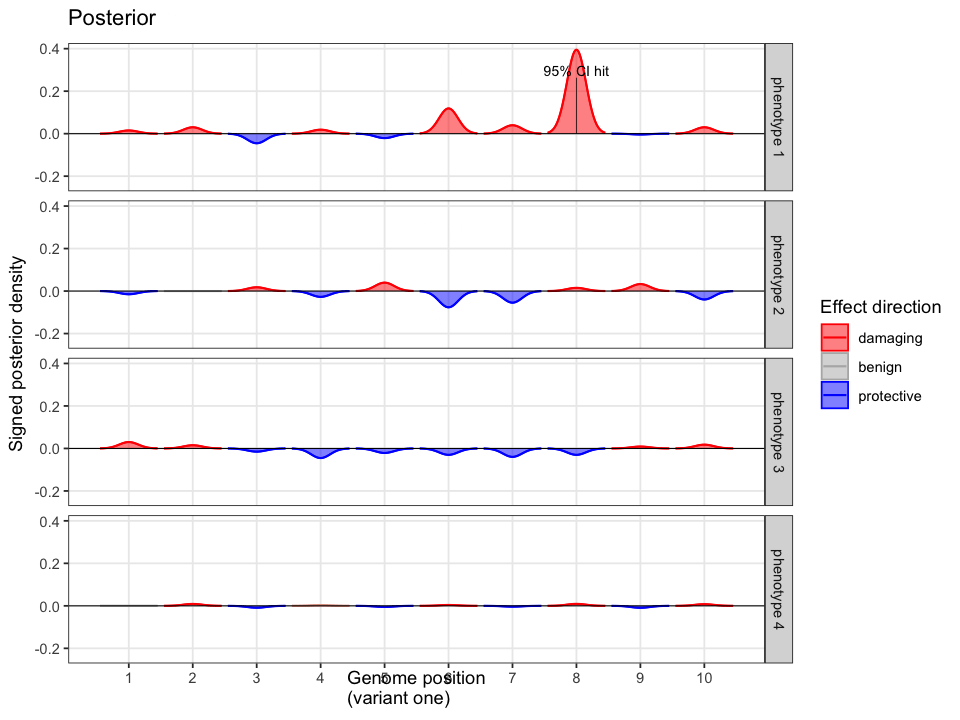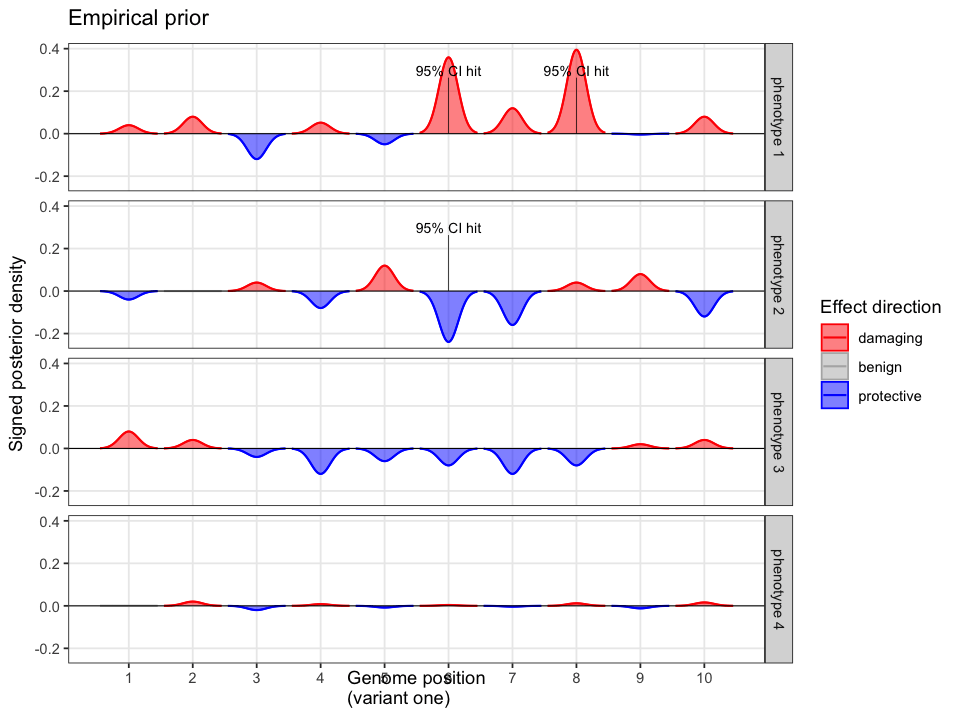
We begin with priors derived from population-scale datasets capturing biological knowledge such as variant frequency, gene and protein function, domain-level constraint, and regional tolerance to variation.
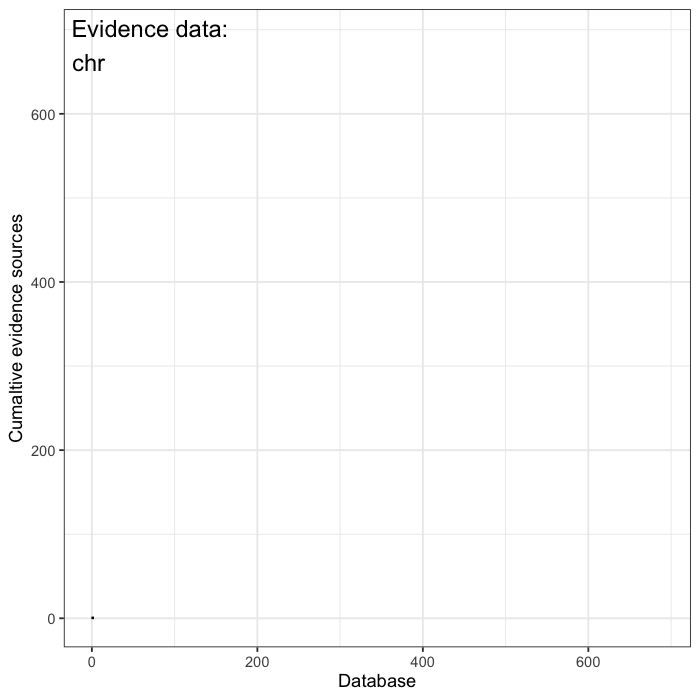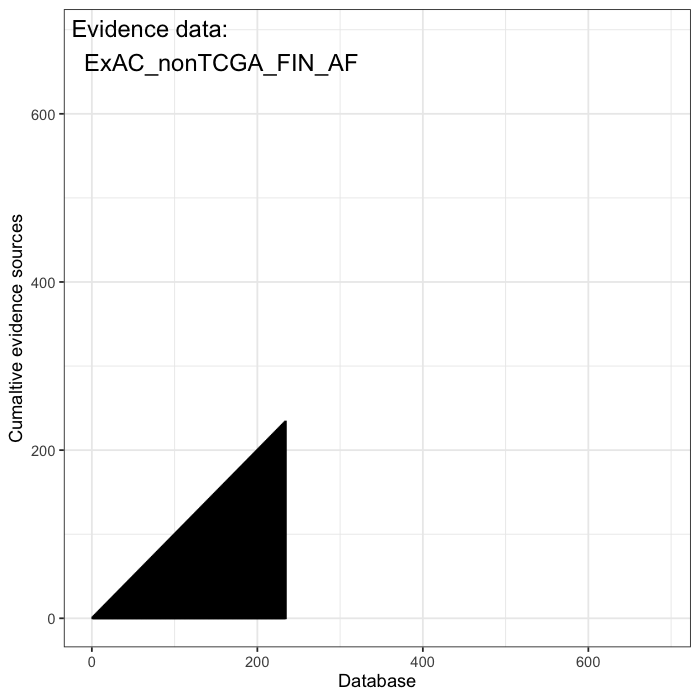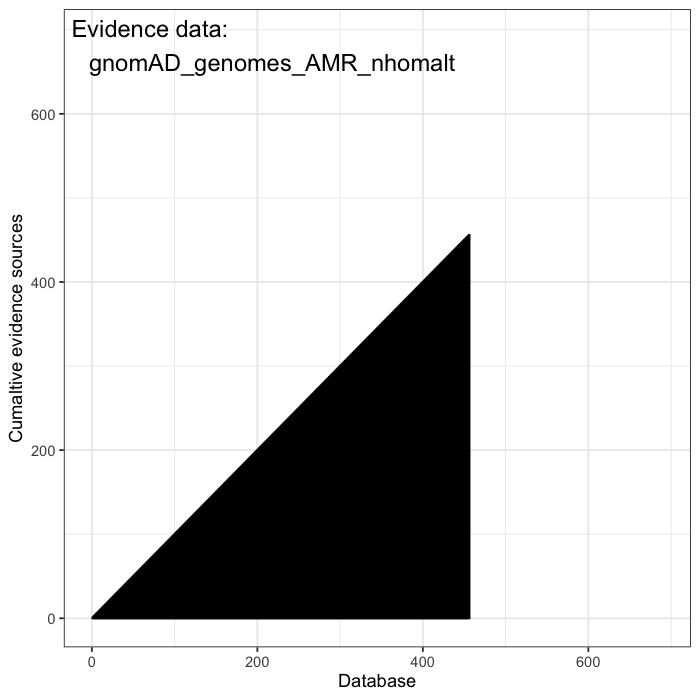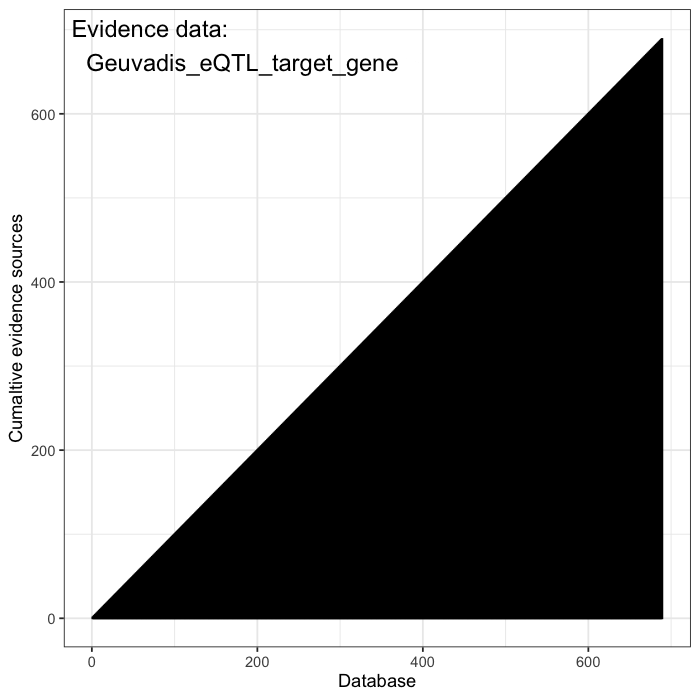
Cumulative evidence from these sources is integrated to estimate the prior probability of occurrence for each variant. Some variants are predicted to be highly damaging based on structural or functional annotations, such as AlphaFold or AlphaMissense. Others are constrained by population frequency, with resources like gnomAD enabling the estimation of how likely a random individual is to carry a given variant. Together, these data define a structured prior over the space of plausible causal alleles and provide the foundation for probabilistic inference.
Rather than limiting inference to variants observed in the patient’s sequencing data, it is important to also model potential false negatives, including plausible causal variants that may be missing due to technical artefacts or incomplete coverage. In this simplified example we can define four variant classes: observed causal, observed non-causal, missing causal, and missing non-causal. Prior probabilities are distributed across these classes based on allele frequency, gene-disease associations, and functional annotation.
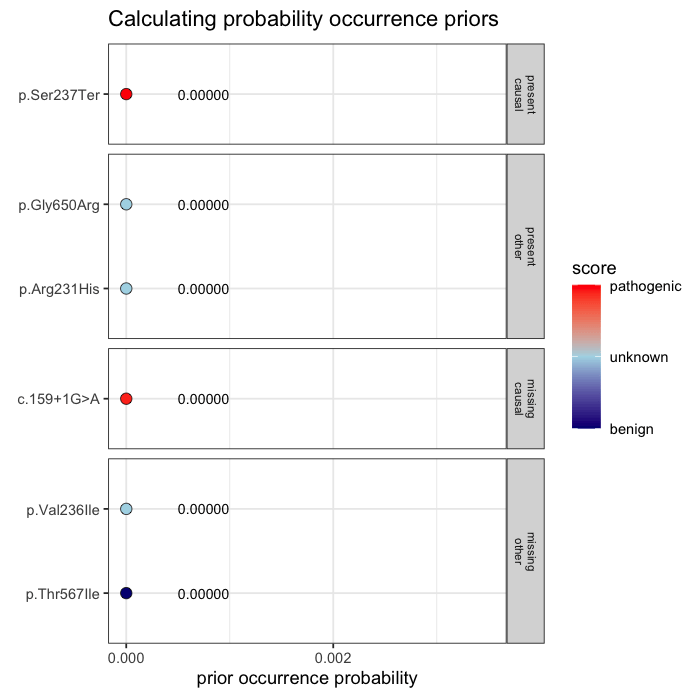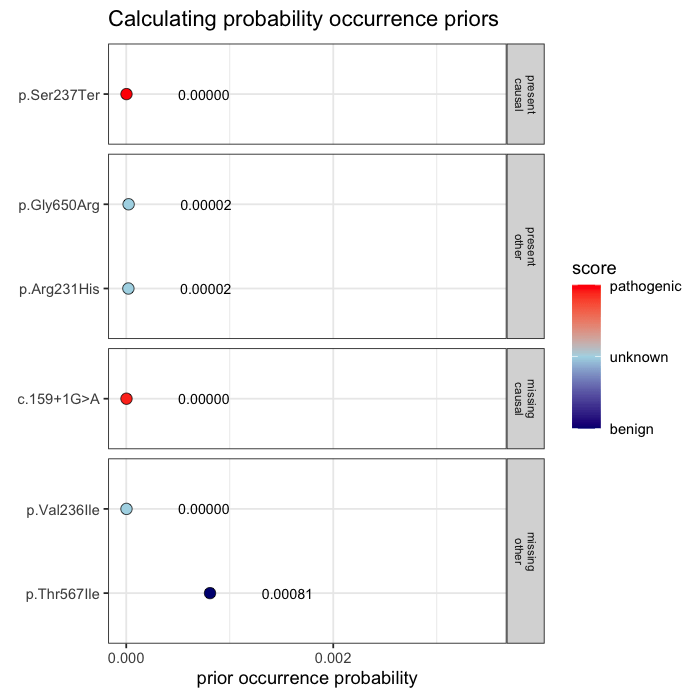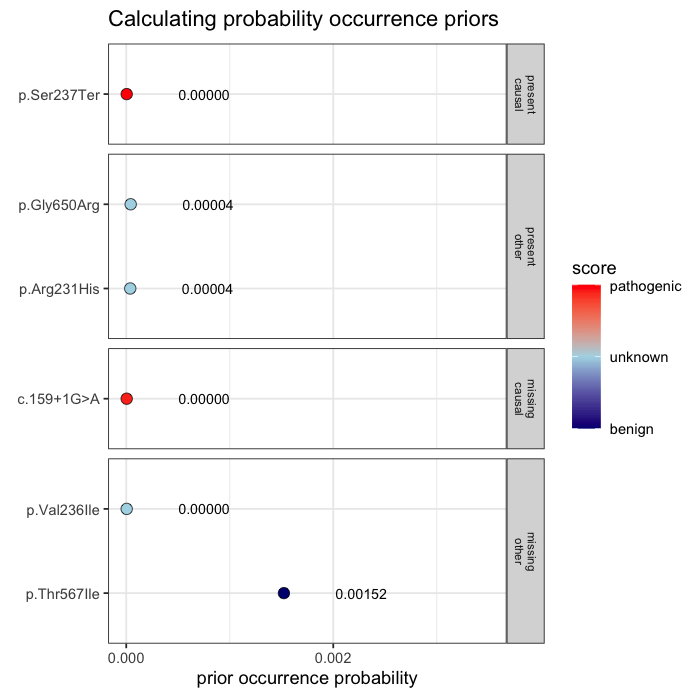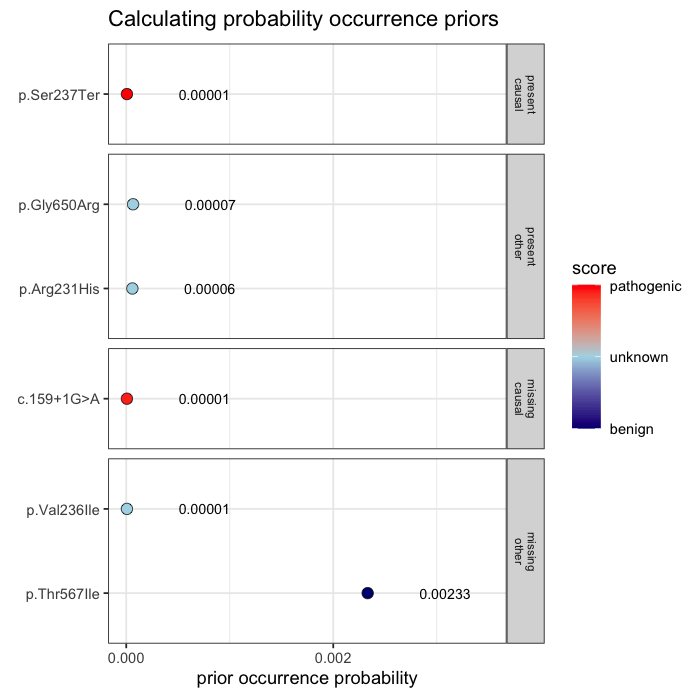
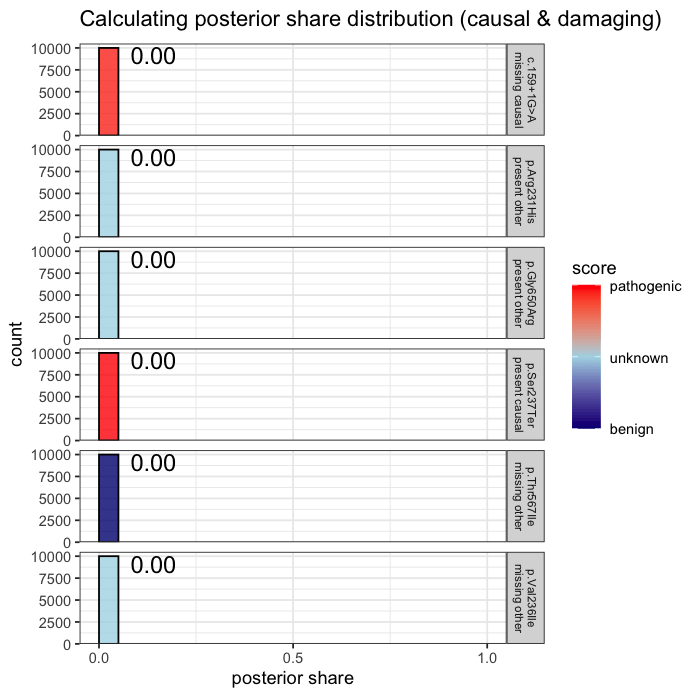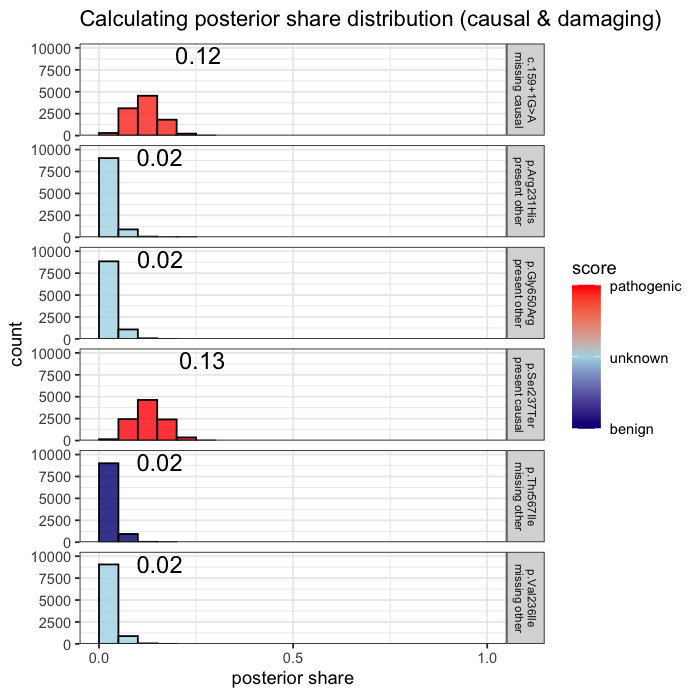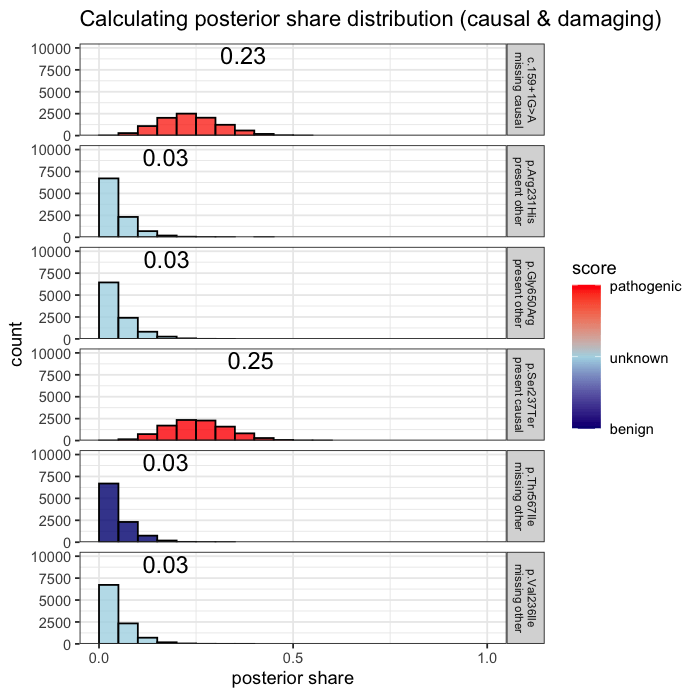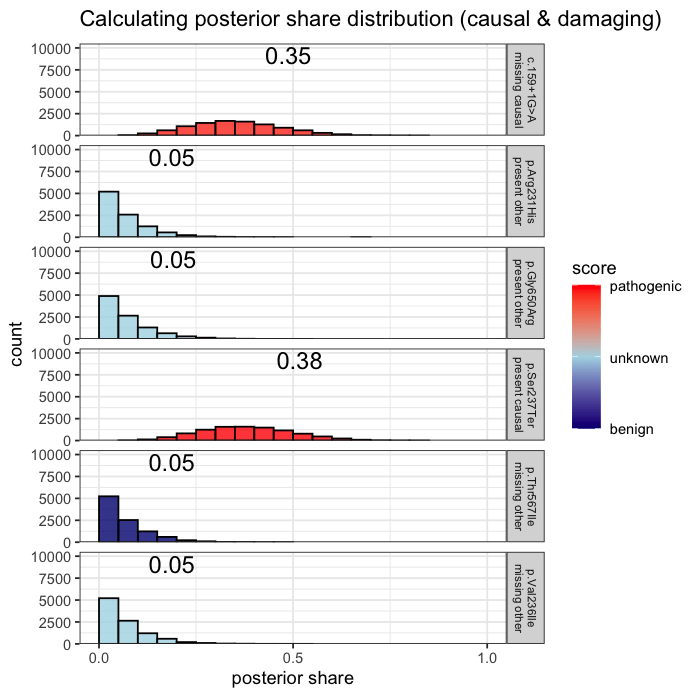
Prior probabilities are computed for all plausible variants, regardless of whether they are ultimately observed in a given patient. This inference step integrates annotation, allele frequency, and gene-phenotype relevance, informed by the patient’s data context (e.g. sequencing scope and phenotype).
Some variants may never appear in the data, but from prior knowledge alone, they remain valid candidates. The model evaluates every plausible variant, not just the ones you can see.
In the example case, two variants dominate the posterior: one observed (p.Ser237Ter) and one unobserved but biologically plausible (c.159+1G>A). The second variant is absent from the patient’s data, but cannot be dismissed, as its pathogenicity and gene context make it a credible candidate. Variants with minimal prior support contribute little to the posterior, but are retained for completeness.
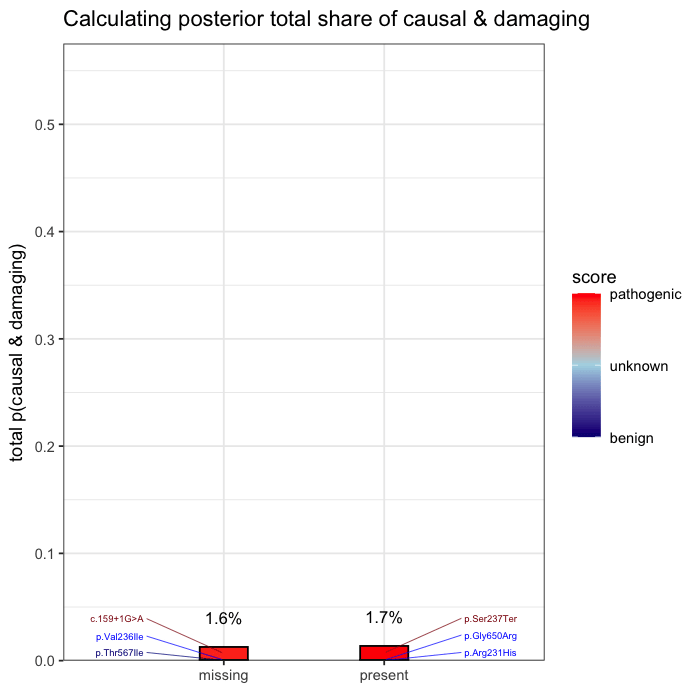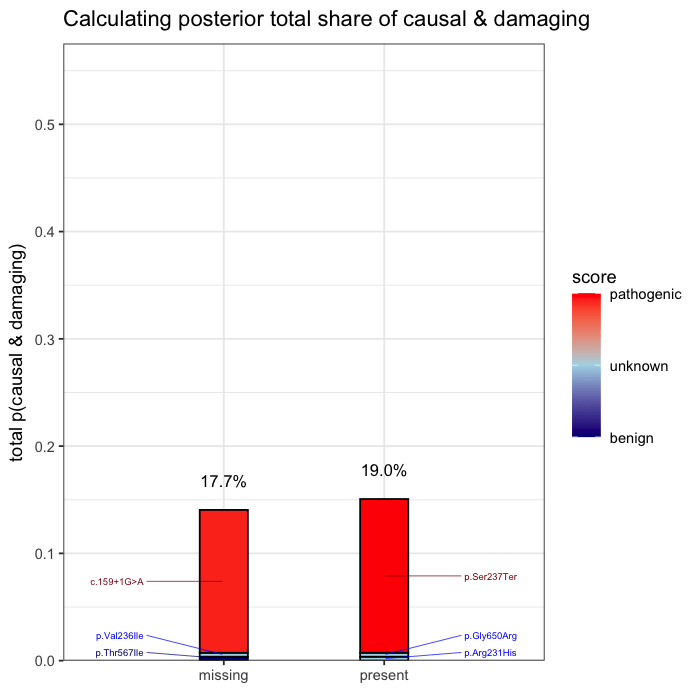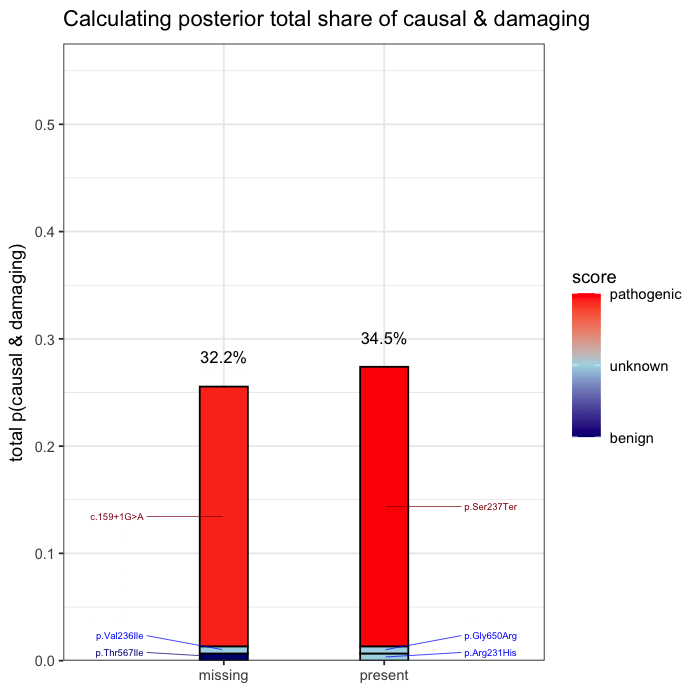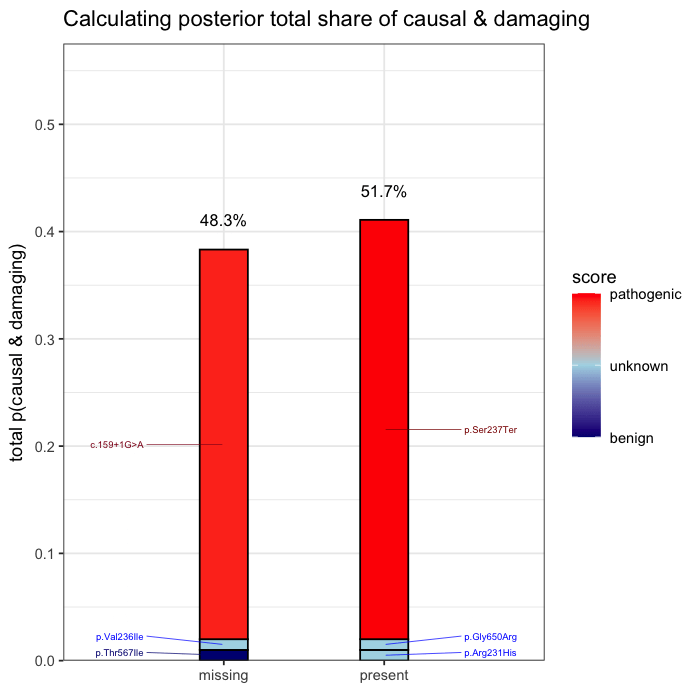
Posterior mass is aggregated by event. All events are independently assessed. The observed variant contributes 43.1% to the posterior probability of disease causation. The other unobserved candidate accounts for the next largest proportion of 40.2%, and the remainder is distributed across low-impact variants. This distribution reflects both statistical uncertainty and the presence of unsequenced positions.
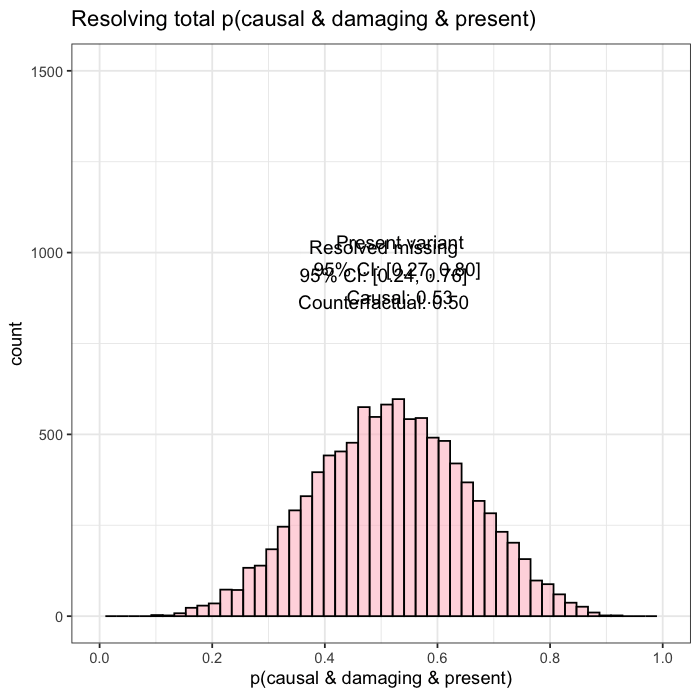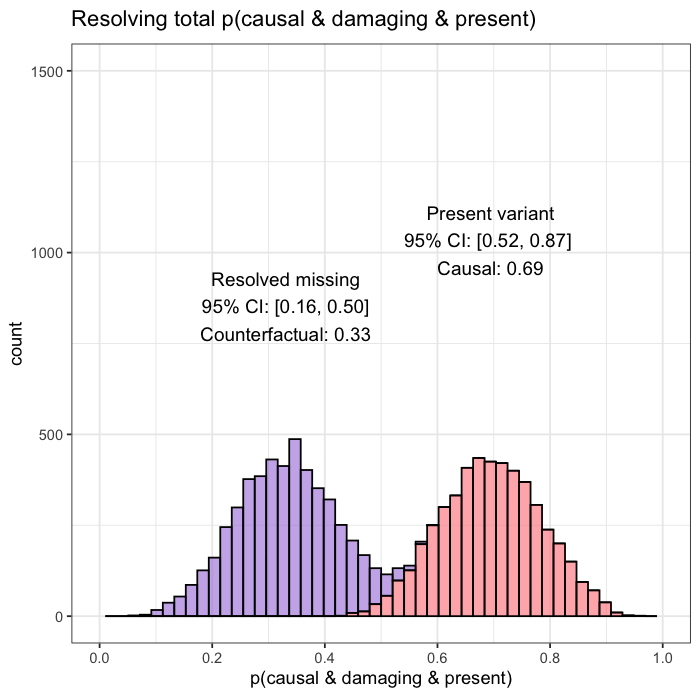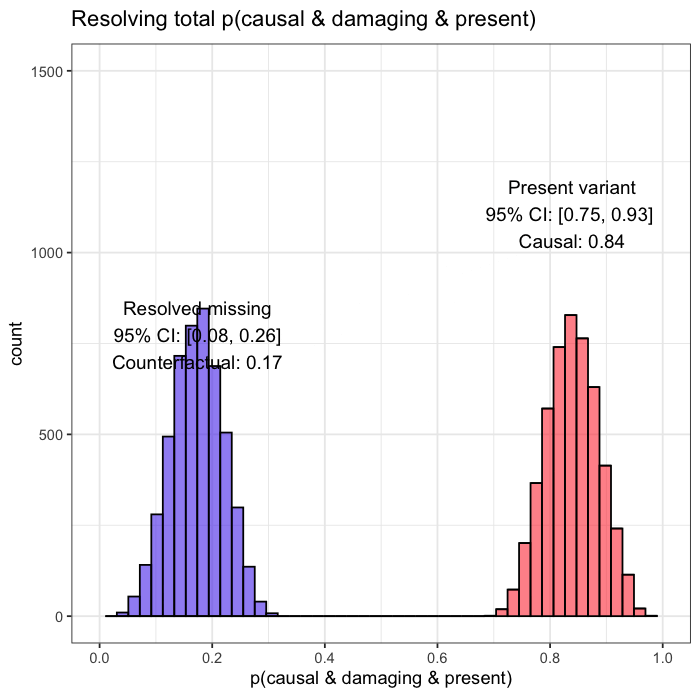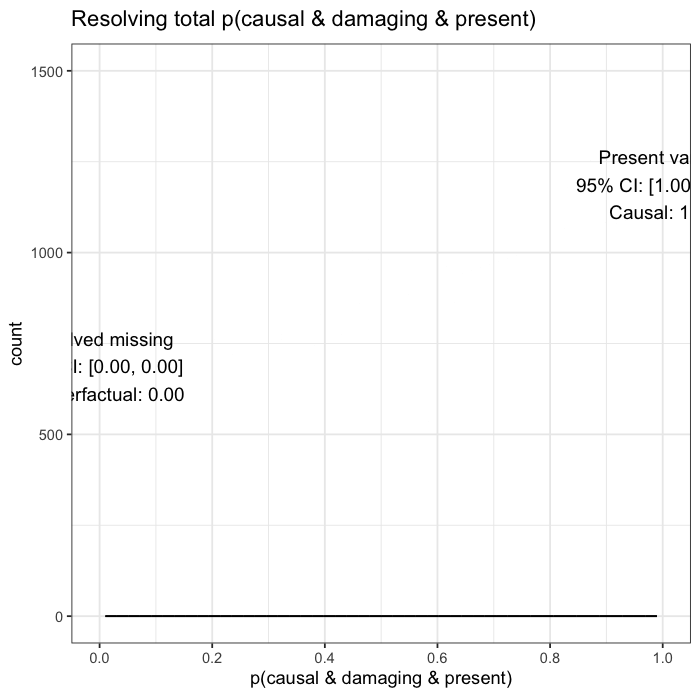
When follow-up analysis confirms that the unobserved candidate is wild-type, the posterior shifts. The probability that the observed variant is causal rises to 77%, with a 95% credible interval of [0.65, 0.90]. The previously unresolved uncertainty is now quantified and updated.
Quant Calc enables variant interpretation through explicit statistical inference. By modelling all potential events, and updating beliefs as new evidence arrives, it delivers rigorous, interpretable conclusions grounded in prior probability and posterior support.
Consider the possibilities of scaling across every combination. A database of empirical priors (like Quant DB) is a powerful tool on its own. In combination with the new layers (using Quant Scan and Calc) we reach a further posterior probability that is conclusive given the peak of our current knowledge.
“This is your last chance. After this, there is no turning back. You take the blue pill - the story ends, you wake up in your bed and believe whatever you want to believe. You take the red pill - you stay in Wonderland, and I show you how deep the rabbit hole goes. Remember, all I’m offering is the truth - nothing more.”
For deeper reading, see:
- Quant: The quantitative omic epidemiology group, et al. “Quantifying prior probabilities for disease-causing variants reveals the top genetic contributors in inborn errors of immunity” medRxiv preprint (2025).
DOI | PDF

