The fastest way to understand a VCF before reading a single variant.
Structured header parsing and standalone HTML reporting for daily review of pipeline outputs.
![]()
VCF headers contain the metadata needed to understand a file before any variant-level interpretation begins. In raw form, that metadata is difficult to inspect quickly, validate consistently, and share with others. Reference assembly, field definitions, filters, samples, and caller-specific tags are present, but not arranged for routine review.
VCFheader does not analyse variants.
It does not replace downstream interpretation tools.
It reads the header only.
It structures the metadata and writes a portable HTML report.
VCF or VCF.gz file → header parse → structured summary and HTML report → faster review, validation, and sharing
When VCFheader is most useful
VCFheader is most useful when a new file arrives and the first task is orientation. It helps users confirm the reference genome, inspect INFO and FORMAT definitions, review filters, check sample content, and identify caller-specific metadata before downstream analysis begins.
It is also useful when outputs need to be tracked across runs, tools, or pipeline versions. A standalone HTML report makes header context easier to review, archive, and share with collaborators.
Installation
Install the released R package from CRAN:
install.packages("vcfheader")
library(vcfheader)
Source: cran.r-project vcfheader
Parse a VCF and write the report
hdr <- parse_vcf_header("file.vcf.gz")
vcfheader(hdr, file = "file_vcfheader.html")
This also works with .vcf files.
You can also let vcfheader() derive the output path from the original input file path:
hdr <- parse_vcf_header("file.vcf.gz")
vcfheader(hdr)
What the report shows
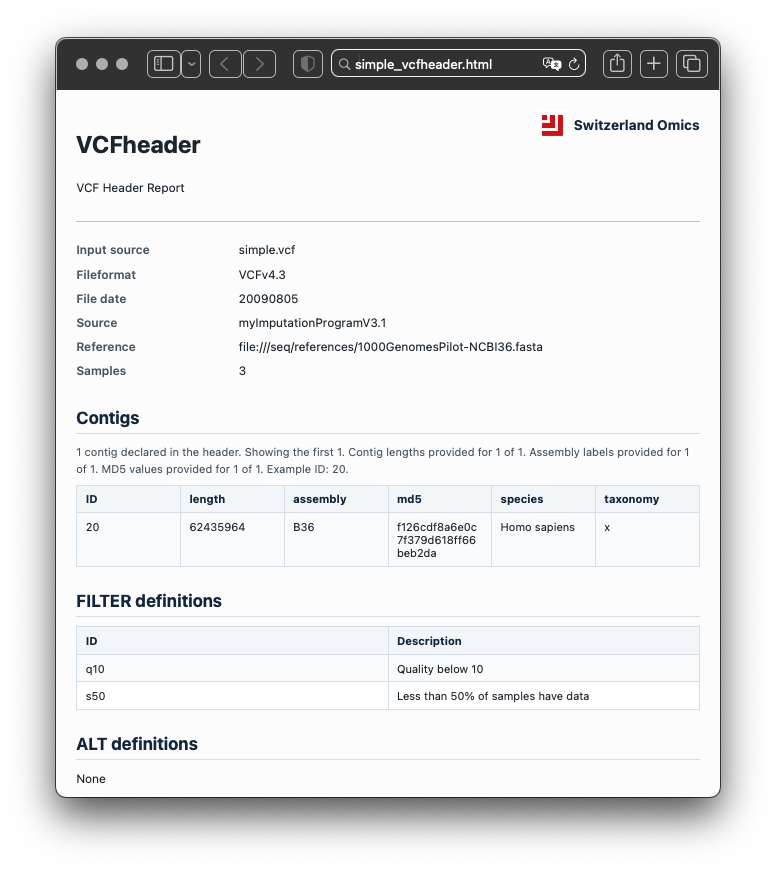
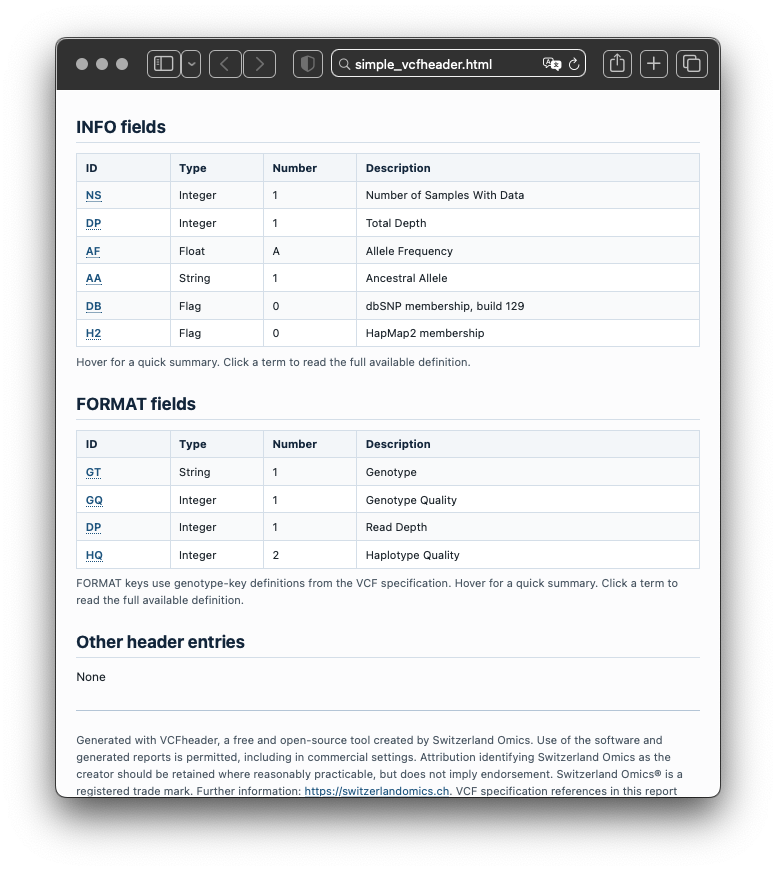
The report summarises file metadata, contigs, INFO fields, FORMAT fields, FILTER definitions, ALT definitions, sample names, warnings, errors, and lightweight inferred annotations. It turns header metadata into a structured record that can be inspected, validated, and shared.
Because only the header is read, review is fast and does not depend on loading full variant records. This makes VCFheader useful for routine checks at the point where a file first enters analysis or review.
Example with bundled files
The package ships with small example files for offline use, including simple.vcf, sv44.vcf, and a prebuilt example report.
library(vcfheader)
simple_vcf <- system.file("extdata", "simple.vcf", package = "vcfheader")
hdr <- parse_vcf_header(simple_vcf)
vcfheader(
hdr,
file = "simple_vcfheader.html"
)
Citation
If you use VCFheader, please cite the software release and package source.
R package
Lawless D. vcfheader: Structured parsing and HTML reporting for VCF headers. Comprehensive R Archive Network.
@Manual{vcfheader_cran,
title = {vcfheader: Fast Genetic Variant Call Format File Header Intelligence and Audit},
author = {Lawless, Dylan},
year = {2026},
note = {R package},
url = {https://CRAN.R-project.org/package=vcfheader}
}
Licence
Generated reports are based on VCFheader, a free and open-source tool created by Switzerland Omics. Use of the software and generated reports is permitted, including in commercial settings, under GPL-3. VCF specification references in generated reports may relate to samtools and the broader HTS specifications ecosystem, distributed under the MIT/Expat Licence by Genome Research Ltd. Source: https://github.com/samtools/samtools. Further reading: https://www.htslib.org/doc/#file-formats.

